MALONYYLIÄ koskevaa
(1) Mitokondriaalinen SIRT4
deasetyloi MCD entsyymin sen K206 -lysiinin ja vaimentaa
entsyymiaktiivisuutta. Entsymin rakenteen lysiinin K206 asetylaatio taas aktivoi sen. Aktivaatiosta taas seuraa AsetylCoA- muodostusta ja hiilidioksidia.
MalonyyliCoA
dekarboksylaasista MDC sitaatti netistä:
MCD vaatii 8 posttranslationaalista modifikaatiota ja niistä viimeinen on lysiinin K472 asetylaatio, mikä aktivoi sen.
SIRTUIINI 4 voi taas mitokondriasssa deasetyloida tämän K472 Ac- ryhmän mikä vaimentaa entsyymin toimintaa.
Entsyymillä kuitenkin on muitakin lokalisoitumisia kuten peroksisomi ja sytoplasma, jossa sillä on lyhemmät isoforminsa toimimassa.
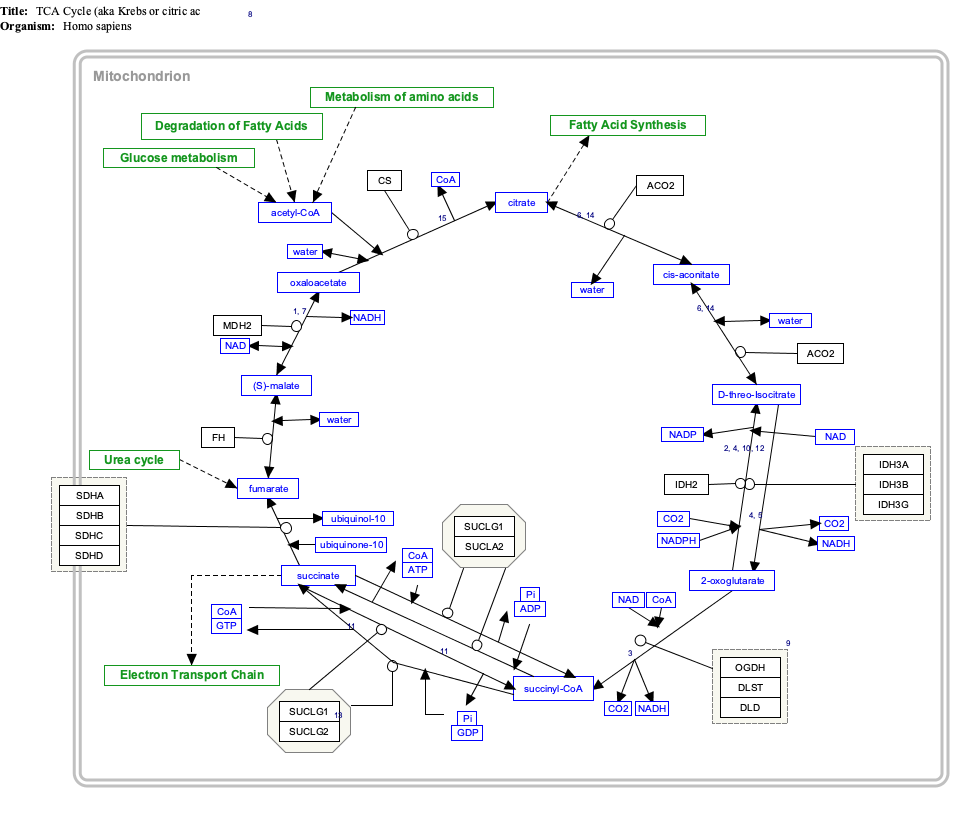
Malonihappoa tuottuu peroksisomeissa parittomien pitkien dihappojen oksidaatiosta päätevaiheessa, (mutta suurin osa menee kuitenkin lopulta suksinyylitiehen sitruunahappokiertoon eikä malonyylitiehen). Siis vain osa menee propionyylivaiheesta akrylyyli CoA- muodon kautta , sitten tulee 3-OH-propionihappoa. sitten malonyylisemialdehydiä ja siitä MDC-entsyymillä asetylCoA ja hiilidioksidia.
MalonylCoA Decarboxylase MCD
Malonyl-CoA decarboxylase (which can also be
called
MCD and
malonyl-CoA carboxyl-lyase) is found
from bacteria to humans, has important roles in regulating fatty acid
metabolism and food intake, and it is an attractive target for drug
discovery. It is an enzyme associated with
Malonyl-CoA
decarboxylase deficiency. In humans, it is encoded by the MLYCD
gene.
MCD presents two
isoforms
which can be transcribed form one gene: a long isoform (54kDa),
distributed in mitochondria, and a short isoform (49kDa) that can be
found in peroxisomes and cytosol. The long isoform includes a
sequence of signaling towards mitochondria in the N-terminus; whereas
the short one only contains the typical sequence of peroxisomal
signaling PTS1 in the C-terminus, also shared by the long isoform.
MCD is a protein
tetramer,
an
oligomer
formed by a dimer of heterodimers related by an axis of binary
symmetry with a rotation angle of about 180 degrees. The strong
structural asymmetry between the monomers of the heterodimer suggests
a half of the sites reactivity, in which only half of the active
sites are functional simultaneously. Each monomer contains basically
two domains:
-
The
N-terminus
one, which is involved in oligomerization and has a helical
structure of eight helixes organised as a bundle of four
antiparallel helixes with two pairs of inserted helixes.
-
The
C-terminus
one is where malonyl-CoA catalysis takes place and which is present
in GCN5- Histone acetyiltranferase family. It also includes a
cluster of seven helixes.
However, the binding site for malonyl-CoA in MCD
presents a variation with respect to their homologous: the center of
the binding site has a glutamic residue instead of a glycine, acting
as a molecular lever in the substrate releasing.
As said before, MCD presents a half of the sites
reactivity, due to the fact that each
heterodimer
has two different structural conformations: B state (bound), in which
the substrate is united; and U conformation (unbound), where the
substrate union isn't allowed. According to this, the half of the
sites mechanism might present a consumption of catalytic energy.
Nevertheless, the conformational change produced in a
subunit
when changing from the B state to the U state (which produces the
release of the product) coincides with the formation of a new union
site in the
active
site of the neighbour subunit when changing from the U stat to B
state. As a result, the conformational changes synchronised in the
pair of subunits facilitates the
catalysis
despite the reduction of the number of available active sites.
Each monomer of that structure exhibits a large
hydrophobic interface with the possibility to form an inter subunit
disulfide bridge. Heterodimers are also interconnected by a small
C-terminus domain interface, where a pair of cysteines is properly
disposed. The disulfide bonds gives to MCD the capability to form a
tetrameric enzyme linked by inter subunits covalent bonds in the
presence of oxidants such as hydrogen peroxide.
-
Processing and
post-translational modifications
Malonyl-CoA decarboxylase is firstly processed as a
pro-protein
or
proenzyme, in
which the
transit
peptide, whose role is to transport the enzyme to a specific
organelle (in this case the mitochondria), comprises the first 39
amino acids (beginning with a
methionine
and ending with an
alanine).
The polypeptide chain in the mature protein is comprised between
amino acid 40 and 493.
In order to turn into an active enzyme, MCD undergoes 8
post-translational
modifications (PTM) in different amino acids.
The last one,
which consists of an acetylation
in the amino acid lysine
in position 472, activates malonyl-CoA decarboxylase activity.
Similarly, a deacetylation
in this specific amino acid by SIRT4
(a mitochondrial protein) represses the enzyme activity.
Another important PTM is the formation of an interchain
disulfide bond
in the amino acid
cysteine
in position 206, which may take place in peroxisomes, as the
cytosolic and mitochondrial environments are too reducing for this
proces
MLYCD is strongly expressed in heart, liver and some other tissues
like kidney. This gene is also weakly expressed in many other tissues
such as brain, placenta, testis, etc.
[1][4][5]
The
enzyme
malonyl-CoA decarboxylase (MCD) functions as an indirect via of
conversion from malonic semi aldehyde to
acetyl-CoA
in
peroxisomes.
This is due to the fact that the
beta
oxidation of long chain
fatty
acids with an odd number of carbons produces
propionyl-CoA.
Most part of this metabolite is transformed into
succinyl-CoA,
which is the precursor of the
tricarboxylic
acid cycle. The major alternative route by which the
propionyl-CoA is metabolized is based on its conversion to
acrylyl-CoA. After that, it is converted to 3-hydroxy propionic acid
and finally to malonic semi-aldehyde. As soon as malonic semi
aldehyde is produced, it is indirectly transformed into acetyl-CoA.
This conversion has been
detected only in bacteria,
[6]
in the other natural kingdoms there is no scientific evidence to
prove it.
[7]
Malonyl-CoA
is an important
metabolite
in some parts of the cell.
In peroxisomes, the accumulation of
this substance causes
malonic
aciduria, a highly pathogenic disease. To avoid it malonyl-CoA
decarboxylase (MCD) converts malonyl-CoA into acetyl-CoA through the
following reaction:
In peroxisomes, it is proposed that this enzyme
could be involved in degrading intraperoxisomal malonyl-CoA, which is
produced by the peroxisomal beta oxidation of
odd chain length
dicarboxylic
fatty acids (odd chain length DFAs). While long and medium
chain fatty acids are oxidized mainly in the mitochondria,
DFAs
are oxidized primarily in peroxisomes, which degrade
DFAs
completely to malonyl-CoA (in the case of odd chain length DFAs)
and
oxalyl-CoA
(for even chain length DFAs). The
peroxisomal form of MCD
could function to eliminate this final malonyl-CoA.
In the cytosol, malonyl-CoA can inhibit the
entrance of fatty acids into the mitochondria and it can also act as
a precursor for the fatty acids synthesis.
Cytoplasmic MCD is
thought to play a role
in the regulation of cytoplasmic malonyl-CoA
abundance and,
therefore, of mitochondrial fatty acid uptake and
oxidation.
[8]
It has been observed that MCD
mRNA
is most abundant in
cardiac and
skeletal muscles, tissues in which
cytoplasmic malonyl-CoA is a strong inhibitor of mitochondrial fatty
acid oxidation and which derive significant amounts of energy from
fatty acid oxidation.
Malonyl-CoA also plays
an
important role inside the mitochondria, where it is an
intermediary between fatty acids and acetyl-CoA,
which will be a
reserve for the
Krebs
cycle.
When Malonyl-CoA acts as an intermediary between fatty acids
and acetyl-CoA in the mitochondria, mitochodnrial MCD is believed to
participate in the elimination of the residual malonyl-CoA, so that
acetyl-CoA can enter the Krebs cycle.
MCD also plays a role in the regulation of
glucose
and
lipids as fuels
in human tissues.
Malonyl-CoA concentrations are crucial in the
intracellular energetic regulation and the
production or
degradation of this metabolite delimits the use of glucose or lipids
to produce
ATP.
The diseases related with MCD can be caused by its
mislocalization, mutations affecting the gene MLYCD, its accumulation
in peroxisomes and, mainly, its deficiency.
MCS deficiency is a rare autosomal disorder that
is widely diagnosed by neonatal screening and it is caused by
mutations in MLYCD. It causes many symptoms: brain abnormalities,
mild mental retardation, seizures, hypotonia, metabolic acidosis,
vomiting, excretion of malonic and methylmalonic acids in urine,
cardiomyopathies, and hypoglycemia. More rarely, it can cause
rheumatoid arthritis too.
In peroxisomes, the accumulation of MCD substance also
causes pathological symptoms, which are similar to MCS deficiency:
malonic aciduria, a lethal disease in which patients (normally
children) have delayed development and can suffer from seizures,
diarrhoea, hypoglycaemia and cardiomyopathy, as well.
Others symptoms caused by an
altered action of MCD can be abdominal pain and chronic
constipation.
[9]
Because the formation of
interchain
disulfide
bonds leads to positive
cooperativity
between active sites and increases the affinity for malonyl-CoA and
the catalytic efficiency (in vitro), MCD activity doesn't need the
intervention of any
cofactors
or divalent metal ions.
[13]
MCD is involved in regulating cardiac malonyl-CoA
levels, inhibition of MCD can limit rates of fatty acid oxidation,
leading to a secondary increase in glucose oxidation associated with
an improvement in the functional recovery of the heart during
ischaemia/
reperfusion
injury. MCD is a
potential novel target for cancer treatment.

{kind=link}